
Abstract
Plant species, including algae and fungi, are based on type specimens to which the name of a taxon is permanently attached. Applying a scientific name to any specimen therefore requires demonstrating correspondence between the type and that specimen. Traditionally, identifications are based on morpho-anatomical characters, but recently systematists are using DNA sequence data. These studies are flawed if the DNA is isolated from misidentified modern specimens. We propose a genome-based solution. Using 4 × 4 mm2 of material from type specimens, we assembled 14 plastid and 15 mitochondrial genomes attributed to the red algae Pyropia perforata, Py. fucicola and Py. kanakaensis. The chloroplast genomes were fairly conserved, but the mitochondrial genomes differed significantly among populations in content and length. Complete genomes are attainable from 19th and early 20th century type specimens; this validates the effort and cost of their curation as well as supports the practice of the type method.
Similar content being viewed by others

Introduction
The correct application of 18th, 19th and early 20th century plant names to modern specimens is a challenging undertaking. Plant names, including algae and fungi1, are based on type specimens, the original specimens on which species names are based. These specimens are housed in approximately 3,400 official herbaria and maintained by more than 10,000 herbarium curators at museums and universities around the world2. Historically, to assign the correct names to modern collections, type specimens were borrowed for anatomical and morphological comparison. This approach however is fraught with problems, particularly for morphologically simple and/or variable species, e.g., most algae, fungi and numerous land plants, or where type material is missing, fragmented, or lacks the vegetative, reproductive, or geographic information necessary for correspondence with modern collections. Compounding the problem is that many herbarium curators are reluctant and sometimes hostile, to loan material for what is termed “destructive sampling”, the extraction of DNA from a fragment of a type specimen. One of the currently accepted answers to this problem is to collect fresh specimens and perform phylogenetic analyses using standard species markers3,4,5. Another is to use modern DNA to develop representative barcodes of species5,6. The fundamental idea of the barcode is to create a database of comparable sequences that are used by researchers for species determination. A global Barcode of Life Database (BOLD) focusing on the barcode as well as the various online repositories (EMBL, GenBank, DDBJ) contain millions of submissions that serve this purpose. The major problem with these two approaches is the assumption that a barcode from any specimen said to be a particular species is truly representative of the type material of that species. The only indisputable method for linking a species name to type material is by sequencing type specimens7,8,9,10. This approach too has limitations. Specifically, usually only small (~200 base pairs) hypervariable regions of DNA can be obtained11 and therefore complete gene sequences required for phylogenetic analyses are not achievable. The age-old question still remains, how do scientists unite the alpha system of taxonomy to modern systematics?
To address this question we isolated DNA from small herbarium fragments (4 × 4 mm2) of species in the economically important red algal genus Pyropia (Py.), recently segregated from Porphyra (Po.)12 and both marketed as nori as follows: 6 type specimens attributed to Py. perforata (J. Agardh) S.C. Lindstrom, 6 non-type specimens of Py. perforata distributed in the northeast Pacific from Washington to Baja California Sur, Mexico, 1 specimen from the type sheet of Py. perforata attributed to Py. kanakaensis (Mumford) S.C. Lindstrom and the holotype collections of 2 northeastern Pacific species, Py. fucicola (V. Krishnamurthy) S.C. Lindstrom and P. kanakaensis (Fig. 1) (Table 1). The specimens ranged in age from 140 years old (collected in 1874) to recent (collected fresh). Included in this analysis are the type specimens of two species (Po.perforata f. segregata Setchell and Hus and Po. sanjuanensis V. Krishnamurthy (Fig. 1)) considered distinct by some authors13,14 and conspecific with Py. perforata by others15,16.

Images of six type specimens analyzed in this study and their high sensitivity quantitations.
(a), (d), Lectotype (LD-Ag 13037) of Porphyra perforata J. Agardh with 860.75 pg/ul at 98 bp. (b), (e), Holotype (VK-11-00061) of Po. sanjuanensis V. Krishnamurthy with 218.65 pg/ul at 74 bp. (c), (f), Syntype (UC 807662) of Po. perforata f. segregata Setchell & Hus with 3496.84 pg/ul measuring at 100 bp. (g), (j), Lower specimen on the lectotype sheet of Po. perforata, identified as Po. kanakaensis by Conway29 with 5.21 pg/ul at 50 bp (LD-Ag 13038). (h), (k), Holotype (VK-11-00121) of Po. fucicola V. Krishnamurthy with 50.00 pg/ul at 150 bp. (i), (l), Holotype (Mumford #161) of Po. kanakaensis Mumford 24.10 pg/ul at 142 bp. FU = florescence units, bp = base pairs.
Results
Quantitation and Data
High sensitivity quantitation of the DNA extractions indicated intact DNA fragments 35–500 base pairs in length (Fig. 1), with considerable variation in concentration between specimens (e.g. refer to Fig. 1f syntype material of Po.perforata f. segregata and Fig. 1j lower specimen on the lectotype sheet of Py. perforata). Based on the fragmented nature of the DNA, the specimens were subjected to single end 36 bp Illumina next generation sequencing17. The number of filtered sequencing reads generated from the 15 specimens varied from 4,716,038 to 68,784,178 (Table 2). The reads were sufficient to assemble the complete chloroplast genomes from 12 of the 15 specimens and the complete mitochondrial genomes for all 15 of the specimens, with the average N50 for all 15 specimens calculated to 25,274 bp and the average maximum contig length to 54,472 bp (Table 2). Prior to analyzing all of the specimens, filtered reads from the first three type materials (LD-Ag 13037, UC 807662, VK-11-00061) were analyzed for bacterial and human contamination and found to contain less than 0.75% contamination18.
Chloroplast Genome Analysis
The chloroplast genomes of Py. perforata were similar in length (189,752 bp to 189,889 bp), content and gene synteny, all containing 209 protein-coding genes (including 24 ycf and 27 Open Reading Frames (ORFs)), 35 tRNA, 3 ribosomal RNA, totaling 247 genes (Supplementary Figures 1–5, Supplementary Table 1). The partial chloroplast genomes of Py. fucicola and Py. kanakaensis we generated account for 97.5% of the estimated complete genome length. The assembly methods we employed for these two holotypes were unable to resolve a region approximately 4.8 kb in length representing non-identical ribosomal 16S, 23S and 5S repeats. The content and synteny of Py. fucicola and Py. kanakaensis are similar to Py. perforata and other Pyropia species.
Within populations of Py. perforata the chloroplast sequences were highly conserved. Two syntype collections of Po.perforata f. segregata from La Jolla, California were nearly identical (differing by 1 SNP), as were two specimens from the lectotype sheet of Py. perforata from San Francisco, California (6 SNPs, 4 gaps) and two syntype specimens of Py. perforata from Santa Barbara, California (4 SNPs). Comparison of genomes between the type collections of Po.perforata f. segregata and Po.sanjuanensis, differed from the lectotype of Py. perforata by 185 SNPs (+14 gaps) and 75 SNPs (+1 gap), respectively. The non-type material of Py. perforata from Punta San Roque, Baja California Sur showed the greatest amount of intraspecific sequence divergence from Py. perforata, 1,072 SNPs and 75 gaps. Pairwise distances between specimens of Py. perforata ranged from 0.0000–0.0053 (Supplementary Table 2). Interspecific distances between Py. perforata and Py. haitanensis were lowest (0.1178) and highest between Py. perforata and Py. fucicola (0.1453).
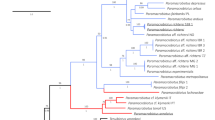
Maximum likelihood analysis of the chloroplast genomes of 18 complete sequences indicates strong support for a clade containing Py. perforata in a sister relationship to Py. haitanensis and Py. kanakaensis (Fig. 2). The same relationships, but with less bootstrap support, were found when a likelihood analysis was performed using only the rbcL gene from the same specimens (Fig. 2). Locally collinear blocks (LCBs) analysis of 12 chloroplast sequences against the published genomes of Pyropia (Py. yezoensis and Py. haitanensis)19 and Porphyra (Po. purpurea and Po. umbilicalis)20,21 identified 33 conserved gene regions using Cyanidiumcaldarium22 as an outgroup. The data confirm that genome structure is highly conserved within the Bangiaceae (Fig. 3). The only apparent difference is that all specimens of Py. perforata contained three fewer non-identical ribosomal 16S, 23S and 5S repeats (approximately 4.8 kb) compared to other Bangiaceae.

Maximum likelihood analysis of chloroplast genomes (a), rbcL sequences (b), mitochondrial genomes (c) and CO1 sequences (d) of Pyropia and Porphyra.
Numbers above branches are maximum likelihood bootstrap values based on 1,000 replicates. The legend below represents the scale for nucleotide substitutions. The analysis was performed using RAxML and the default parameters in Galaxy43,44,45. The tree was constructed with TreeDyn 198.3 at Phylogeny.fr46.

Locally collinear blocks (LCBs) analysis for 17 complete chloroplast genomes.
The figure depicts linearized alignments identifying 33 conserved gene regions for 14 specimens of Pyropia (a–n), two of Porphyra (o–p) and outgrouped with Cyanidiumcaldarium (q). Each chromosome is oriented horizontally and homologous blocks are shown as identically colored regions linked across genomes. Regions that are inverted relative to Py. perforata are shifted below the genome's center axis. Sequence similarities within an LCB are proportional to the heights of interior colored bars. Large sections of white within blocks and gaps between blocks, indicate lineage specific sequences. The analysis shows that chloroplast genomes of Pyropia and Porphyra are similar in content and gene synteny. The single observable difference is the presence of ribosomal 16S, 23S and 5S repeats found in other Pyropia and the two species of Porphyra, but absent in Py. perforata. (a–l), Py. perforata, (a), LD-Ag 13037 (b), VK-11-00061 (c), UC 807662 (d), LD-Ag 13038 (e), UC 95739 (f), LD-Ag 13031 (g), UC 95735 (h), LD-Ag 13032 (i), UC 1450590 (j), UC 2019900 (k), UC 2019901 (l), UC 2019902 (m), Py. haitanensis (n), Py. yezoensis (o), Po. purpurea (p), Po. umbilicalis (q), Cyanidiumcaldarium. The figure was drawn using Mauve 2.3.139.
Mitochondrial Genome Analysis
The mitochondrial genomes of specimens attributed to Py. perforata harbored 55 to 59 genes, with lengths ranging from 32,491 bp (Py. perforata from Carmel, California) to 40,042 bp (holotype of Po. sanjuanensis from San Juan Island, Washington) (Table 2, Supplementary Table 3). Specimens of Py. perforata contained 2–3 ribosomal RNA genes [1–2 large subunit (rnl), 1 small subunit (rns)], 23–24 transfer RNAs, 4 ribosomal proteins, 2 ymfs and 18–19 genes involved in electron transport and oxidative phosphorylation. The number of ORFs varied between specimens (3 ORFs in Py. perforata from Carmel, California to 7 ORFs in the holotype of Po. sanjuanensis) (Supplementary Figures 6–17, Supplementary Table 3). The genome content of Py. fucicola was similar to Py. perforata, however Py. kanakaensis lacked orf546, but contained orf729.
The mitochondrial genome sequences within populations of Py. perforata were similar. Two syntype collections of Po.perforata f. segregata from La Jolla, California were nearly identical (differing by 5 SNPs, 2 gaps), as were the two specimens from the lectotype sheet of Po. perforata from San Francisco, California (4 SNPs, 2 gaps) and two syntype specimens of Py. perforata from Santa Barbara, California (3 SNPs). In contrast, the genomes of Py. perforata from different populations varied in their content and length. The type collections of Po.perforata f. segregata and Po.sanjuanensis differed from the lectotype of Py. perforata by 120 SNPs (+8 single nucleotide gaps and 3 large gaps) and 106 SNPs (+3 single nucleotide gaps and 3 large gaps), respectively. The specimen from Punta San Roque, Baja California Sur exhibited the greatest intraspecific variation compared to the lectotype of Py. perforata, showing 934 SNPs, 127 single/multiple length gaps and 1 large gap. Pairwise distances between specimens of Py. perforata ranged from 0.0000–0.0641 (Supplementary Table 4). Distances between the holotype of Py. kanakaensis and a more recent collection of this species from Land's End, San Francisco was 0.0039. Interspecific distances between Py. perforata and Py. fucicola were lowest (0.1963) and highest between Py. perforata and Py. yezoensis (0.3226). Distances between Py. yezoensis, Py. haitanensis, Py. kanakaensis and Py. fucicola ranged from 0.1113–0.3499.
Maximum likelihood analysis of the complete mitochondrial genomes found strong support for a single monophyletic clade containing Py. perforata, which was sister is position to Py. haitanensis and Py. kanakaensis (Fig. 2). Phylogenetic analysis of the same representatives using only their cytochrome oxidase 1 sequences (664 bp) failed to resolve the populations of Py. perforata and found different relationships for the other species of Pyropia (Fig. 2). LCB analysis and linearized barcode alignments of the 15 Pyropia generated here, against those of published Pyropia and Porphyra21,23,24,25,26, identified 18 conserved gene regions (Fig. 4). The alignments depict numerous insertion/deletion events among populations of Py. perforata and between Py. perforata and other species of Pyropia. No alignment differences were observed within populations of Py. perforata, but significant polymorphisms were evident among populations of this species. Barcode findings were similar to those of the LCB analysis (Fig. 5). Most notably the intraspecific mitochondrial genome content differences for Py. perforata were: 1) the lectotype and two other collections of Py. perforata (San Francisco and Baja California Sur) lack the entire 2,326 bp large subunit ribosomal intron present in other species of Pyropia, whereas some Py. perforata and Py. kanakaensis both lack part (1,274 bp) of the same intron, 2) type material of Py. perforata contains a single orf546 gene, whereas the other specimens either have an additional non-identical orf546 repeat totaling 2,478 bp in size, or totally lack orf546 (Carmel, California), 3) Py.perforata from Santa Barbara and La Jolla lack a 2,075 bp open reading frame (orf693) that is present in the other Py. perforata specimens and in other species of Pyropia, 4) Py.perforata from La Jolla, California codes for an additional tRNA (histidine) and 5) the holotype material of Po.sanjuanensis contains a 2,590 bp insertion that codes for a group II intronic open reading frame (orf813) not present in the other Py. perforata, but present in Py. haitanensis and Py. tenera.

Locally collinear blocks (LCBs) analysis for 20 complete mitochondrial genomes.
The figure depicts linearized alignments identifying 18 conserved gene regions for six species of Pyropia (a–r) and two of Porphyra (s–t). Each chromosome is oriented horizontally and homologous blocks are shown as identically colored regions linked across genomes. Regions that are inverted relative to P. perforata are shifted below the genome's center axis. Sequence similarities within an LCB are proportional to the heights of interior colored bars. Large sections of white within blocks and gaps between blocks, indicate lineage specific sequences. The analysis shows that mitochondrial genomes within populations of Py. perforata are similar in content and length, but highly variable between populations and other Pyropia. (a–l), Py. perforata, (a), LD-Ag 13037 (b), LD-Ag 13038 (c), UC 95735 (d), LD-Ag 13031 (e), LD-Ag 13032 (f), UC 2019900 (g), UC 2019901 (h), UC 2019902 (i), UC 1450590 (j), UC 807662 (k), UC 95739 (l), VK-11-00061 (m), Mumford #161 (n), UC 1863890 (o), VK-11-00121 (p), Py. haitanensis (q), Py. yezoensis (r), Py. tenera (s), Po. purpurea (t), Po. umbilicalis. The figure was drawn using Mauve 2.3.139.

Linearized barcode representation of 20 aligned complete mitochondrial genomes for six species of Pyropia (a–r) and two of Porphyra (s–t).
Matching colors between rows represent similar DNA sequences and blanks (white blocks) represent deletion events. The analysis shows that mitochondrial genomes within populations of Py. perforata are similar in content, but highly variable between populations and other Pyropia. Deletions of the two large ribosomal subunit introns (rn1 intron and orf546), a large 2,075 bp ORF (orf693) and the insertion of a 2,590 bp ORF (orf813), as well as the insertion of orf729 distinguish populations and different species of Pyropia. (a–l), Py. perforata, (a), LD-Ag 13038 (b), LD-Ag 13037 (c), UC 1450590 (d), VK-11-00061 (e), LD-Ag 13031 (f), LD-Ag 13032 (g), UC 2019900 (h), UC 2019902 (i), UC 2019901 (j), UC 95739 (k), UC 807662 (l), UC 95735 (m), VK-11-00121 (n), Py. yezoensis (o), Py. tenera (p), UC 1863890 (q), Mumford #161 (r), Py. haitanensis (s), Po. purpurea (t), Po. umbilicalis. The figure was illustrated using Jalview41.
Phylogenetic Markers
Analysis of the standard chloroplast markers ribulose-1,5-bisphosphate carboxylase/oxygenase (rbcL) and the universal plastid amplicon (UPA), plus the universal mitochondrial barcode marker cytochrome oxidase 1 (CO1), found few polymorphisms (Supplementary Table 5) among populations of Py. perforata from Alaska, USA to Baja California Sur, Mexico. The rbcL gene for Py. perforata showed 0–2 (6) bp variation (the 6 bp variation was exhibited solely in the specimen from Baja California Sur) and the lectotype sequence of Py. perforata was identical to three sequences deposited in GenBank from Alaska, USA and British Columbia, Canada; no differences for the UPA gene were observed among Py. perforata populations and all 13 genome sequences matched the two 371 bp sequences deposited in GenBank from British Columbia specimens; no polymorphisms were identified for CO1 between the lectotype and other Py. perforata, with the exception of the Py. perforata from Baja California Sur (which differed by 3 bp) and the holotype specimen of Po. sanjuanensis. The latter was found to contain orf813 inserted in the CO1 gene (Fig. 5). As noted above, this specific orf813 organization is also found in Py. haitanensis and Py. tenera. Comparison of CO1 sequences from the Py. perforata genomes to those in GenBank found 12 exact matches from specimens from British Columbia. Analysis of the holotype of Py. kanakaensis found an exact match in GenBank to the rbcL sequence generated from a specimen from British Columbia and two exact matches for CO1 from specimens of Py. kanakaensis from the same province. The holotype of Py. fucicola failed to exactly match any sequences in GenBank for rbcL and UPA, but its CO1 barcode was identical to seven sequences deposited under the name Py. fucicola from British Columbia.
Discussion
The first plastid and mitochondrial genomes from red algae were determined for Porphyra purpurea20,24. The organellar genomes of other Bangiaceae soon followed19,21,23,25,26. Excluding six red algal florideophyte chloroplast genomes and ten mitochondrial genomes, in total GenBank contains the complete circular genomes of two species of Porphyra (Po. purpurea and Po. umbilicalis), three Pyropia mitochondrial genomes (Py. yezoensis, Py. haitanensis and Py. tenera) and two Pyropia chloroplast genomes (Py. yezoensis, Py.haitanensis). This study investigated genomic divergence at both the intraspecific and interspecific levels to test the current taxonomic classification of Py. perforata. We analyzed the type specimens of Po. perforata f. segregata and Po. sanjuanensis and compared the genetic distances exhibited by these specimens to two closely related species, Py. fucicola against Py. yezoensis12. The distances between the latter were calculated to 0.0338 for the chloroplast genome. The same comparison done for Po.purpurea and Po. umbilicalis, was 0.0833, well within the range observed for all Pyropia distances compared in this study (0.0338–0.1455). The range of divergence between the lectotype of Py. perforata and the types of Po. perforata f. segregata (0.0009) and Po. sanjuanensis (0.0004), fall well within that of all Py. perforata from Washington to Baja California Sur (0.0000–0.0053). It is thus concluded that this variation represents intraspecific variation. Conversely, mitochondrial distances between Py. fucicola and Py. yezoensis, plus Py. fucicola and Py. tenera, were 0.1463 and 0.1113, respectively. Pairwise distances between various Pyropia species were quite high (0.1113–0.3499). For Po.purpurea and Po. umbilicalis that number was 0.1567. The level of variation observed among populations of Py. perforata was 0.0000–0.0641. Compared to the lectotype of Py. perforata, the types of Po. perforata f. segregata (0.0258) and Po. sanjuanensis (0.0224) fall within the observed intraspecific range. Based on these well-defined pairwise distances, the interspecific delineations using complete plastid evidence is likely around 0.025 and higher and for the mitochondrial genome they are at 0.10 and higher.
Analysis of standard markers27 indicates that scant amounts of variation can be obtained through the marker approach compared to the genomic method of analysis. In comparing the chloroplast variation exhibited by the rbcL gene among populations of Py. perforata, we found a mere 0–2 bp divergence, whereas, the genome data for these same specimens displayed 1 SNP-1,072 SNPs and 75 gaps divergence. Interestingly enough, the maximum likelihood analysis of the rbcL data generated a congruent evolutionary hypothesis compared to the genome data phylogeny. The other chloroplast marker, UPA, failed to exhibit any polymorphisms in this species. The CO1 barcode showed 0–3 bp variation, whereas the genome data for these specimens found content, length (32,491 to 40,042 bp) and SNP variation (3 SNPs–934 SNPs, 127 single/multiple length gaps and 1 large gap). These results suggest that the marker based approach to phylogenetics is failing to identify a large amount of cryptic molecular diversity in these algae. Comparison of the CO1 phylogeny to the genome derived tree found incongruency. The CO1 data alone was unable to resolve populations of Py. perforata and supported different relationships compared to the genome-based hypothesis.
All of these results taken together, support previous taxonomic and phylogenetic conclusions regarding the synonymy of the names Po. perforata f. segregata and Po. sanjuanensis under Py. perforata15,16. This species, although quite variable in its mitochondrial sequence between populations, is circumscribed to accommodate monostromatic thalli that inhabit the uppermost intertidal to the lower intertidal, that are variable in color with ruffled margins, vary in thickness from 40–60 μm, are monoecious and reproduce sexually with tiers of zygotosporangia in 2 or 4 (mixed and not mixed with vegetative cells) and spermatangia in tiers of 8, but that also asexually reproduce via aplanospores and show a karyotype of 2 or 313,28,29,30,31,32. One of the specimens that was analyzed, LD-Ag 13038 (Fig. 1g), mounted on the lectotype sheet of Py. perforata, was previously attributed incorrectly to Py. kanakaensis based on anatomical examination29. This specimen should be designated syntype material, especially in light of the fact that it is excessively perforate and the sheet carries the inscription Porphyra perforata in the author's (J. Agardh's) handwriting. The other specimen that was misidentified as Py. perforata (UC 1863890 from Land's End, San Francisco, California), was determined by mitochondrial genome and partial plastid analysis to be assignable to Py. kanakaensis.
Worldwide herbaria are estimated to contain 300 million specimens and nearly all of them are not being used for molecular phylogenetic studies. Of the estimated 70,000 plant species still to be described, more than half already have been collected and are stored in herbaria33. In an age when administrators of universities are cutting funds or considering closure of herbaria on the grounds of obsolescence, there is a need for a method that will allow for type and non-type specimens to be compared against existing older names, as well as future names. Our data show that this need can be satisfied using very small amounts of archival herbarium tissue. The methodologies used here are optimized for low DNA quality and concentration for library construction (several of the samples contained less than 0.5 ng of total DNA). The amount of material required for this type of analysis is similar to that traditionally used for microscopic examination. In addition, our results show that large amounts of single read sequence data are not required to decipher the chloroplast and mitochondrial genomes. In this case we assembled the two circular genomes of the specimen of Py. perforata from Baja California Sur with only 4,716,038 filtered reads. Once deciphered, the large amount of information housed in the chloroplast and mitochondrial genomes likely eliminates the need for future sampling of the type material for organellar purposes. The complete circular genomes of type specimens can be used in part (i.e. markers) or in total, to address barcode, phylogenetic, conservation, taxonomic, historical, evolutionary and population studies. This data shows that 19th and early 20th century herbarium specimens have great value for current and future systematic and genomic studies and with respect to type specimens, are essential for the accurate application of species names for all plants, algae and fungi where ample material was archived.
Methods
DNA was isolated following the protocol of Lindstrom et al.9, with the following exception: nucleic acids were resuspended with 60 μl of elution buffer. The extractions were performed using 4 × 4 mm2 of material following the precautionary contamination guidelines outlined by Hughey and Gabrielson11. The DNA quality and quantity was analyzed by the High-Throughput Genomics Center (HTGC) on an Agilent 2100 Bioanalyzer™ following the manufacturer's instructions. The genome library was constructed based on a modified TruSeq protocol developed by HTGC (Supplementary Methods). The 36 bp single end sequencing analysis was performed using the manufacturer's protocol via the cBot and HiSeq 2000 by HTGC. Filtered reads were base called using Illumina's standard pipeline, then assembled using the Bio-Linux 734 platform with Velvet35 running on auto settings. After the first run, the data was then rerun optimizing for the expected cutoff and coverage cutoff based on the coverage data from the first assembly. Specimens with more than 15 million reads were assembled using the kmer = 31, while those with less than 8 million were assembled with kmer = 25. The resulting contigs were searched at NCBI using Megablast, then aligned contigs were ordered according to reference sequences (Py. yezoensis, Py. haitanensis and Po. purpurea). To validate the joined contigs, targeted PCR and sequencing and assembly comparisons to Metavelvet36 contig results, were analyzed on the first three genomes assembled (LD-Ag 13037, UC 807662, VK-11-00061). Genomes processed later were confirmed by aligning sequence reads against a draft assembly in NextGENe® (SoftGenetics LLC). The ORFs were annotated using NCBI ORF-finder and alignments obtained via BLASTX and BLASTN searches at NCBI. The tRNAs were identified using the tRNAscan-SE 1.21 web server37 and the rRNAs using the RNAmmer 1.2 server38. LCB alignments were generated using ProgressiveMauve39 with a seed of 21 for the chloroplast and mitochondrial alignments, with the ‘Use seed families’ option selected. The barcode alignment of the mitochondrial data was performed with MAFFT 7.058140 using default settings and the results were presented with Jalview41. Alignments results from MAFFT were analyzed with RaxML42 using the default parameters in Galaxy43,44,45 and the phylogenetic tree was visualized with TreeDyn 198.3 at Phylogeny.fr46. Pairwise distances were calculated using the default settings (GTR substitution model) by DIVEIN47. Deconseq analysis to determine human and bacterial contaminant percentages was analyzed against the following: Human-Reference GRCh37, 57,317 unique 18S sequences and 2,206 unique bacterial genomes at the 90–94% default settings.
Additional Information
Genbank accession numbers KC904971, KF515971-KF515975, KJ708761-KJ708772, KJ776827-KJ776837
References
McNeill, J. et al. International Code of Botanical Nomenclature For Algae, Fungi and Plants (Melbourne Code): adopted by the Eighteenth International Botanical Congress Melbourne, Australia, July 2011 (Koeltz Scientific Books, 2012).
Staats, M. et al. DNA damage in plant herbarium tissue. PLos ONE 6, e28448 (2011).
Freshwater, D. W. & Rueness, J. Phylogenetic relationships of some European Gelidium (Gelidiales, Rhodophyta) species based on rbcL nucleotide sequence analysis. Phycologia 33, 187–194 (1994).
Presting, G. G. Identification of conserved regions in the plastid genome - implications for DNA barcoding and biological function. Can. J. Bot. 84, 1434–1443 (2006).
Hebert, P. D. N., Cywinska, A., Ball, S. L. & deWaard, J. R. Biological identifications through DNA barcodes. Proc. R. Soc. B. 270, 313–322 (2003).
Saunders, G. W. Applying DNA barcoding to red macroalgae: a preliminary appraisal holds promise for future applications. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 360, 1879–1888 (2005).
Hughey, J. R., Silva, P. C. & Hommersand, M. H. Solving taxonomic and nomenclatural problems in Pacific Gigartinaceae (Rhodophyta) using DNA from type material. J. Phycol. 37, 1091–1109 (2001).
Gabrielson, P. W. Molecular sequencing of Northeast Pacific type material reveals two earlier names for Prionitis lyallii, Prionitis jubata and Prionitis sternbergii, with brief comments on Grateloupia versicolor (Halymeniaceae, Rhodophyta). Phycologia 47, 89–97 (2008).
Lindstrom, S. C., Hughey, J. R. & Martone, P. T. New, resurrected and redefined species of Mastocarpus (Phyllophoraceae, Rhodophyta) from the northeast Pacific. Phycologia 50, 661–683 (2011).
Hind, K. R., Gabrielson, P. W., Lindstrom, S. C. & Martone, P. T. Misleading morphologies and the importance of sequencing type specimens for resolving coralline taxonomy (Corallinales, Rhodophyta): Pachyarthron cretaceum is Corallina officinalis. J. Phycol. 50, in press (2014).
Hughey, J. R. & Gabrielson, P. W. Acquiring DNA sequence data from dried archival red algae (Florideophyceae) for the purpose of applying available names to contemporary genetic species: a critical assessment. Botany 90, 1191–1194 (2012).
Sutherland, J. E. et al. A new look at an ancient order: generic revision of the Bangiales (Rhodophyta). J. Phycol. 47, 1131–1151 (2011).
Krishnamurthy, V. A revision of the species of the algal genus Porphyra occurring on the pacific coast of North America. Pac. Sci. 26, 24–49 (1972).
Conway, E., Mumford, T. F. & Scagel, R. F. The genus Porphyra in British Columbia and Washington. Syesis 8, 185–244 (1975).
Abbott, I. A. & Hollenberg, G. J. Marine Algae of California (Stanford University Press, 1976).
Lindstrom, S. C. & Cole, K. M. An evaluation of species relationships in the Porphyra perforata complex (Bangiales, Rhodophyta) using starch gel electrophoresis. Hydrobiologia 205, 179–183 (1990).
Metzker, M. L. Sequencing technologies – the next generation. Nat. Rev. Genet. 11, 31–46 (2010).
Schmieder, R. & Edwards, R. Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS ONE 6, e17288 (2011).
Wang, L. et al. Complete Sequence and Analysis of Plastid Genomes of Two Economically Important Red Algae: Pyropia haitanensis and Pyropia yezoensis. PLoS ONE 8, e65902 (2013).
Reith, M. & Munholland, J. Complete nucleotide sequence of the Porphyra purpurea chloroplast genome. Plant Mol. Biol. Rep. 13, 333–335 (1995).
Smith, D. R., Hua, J., Lee, R. W. & Keeling, P. J. Relative rates of evolution among the three genetic compartments of the red alga Porphyra differ from those of green plants and do not correlate with genome architecture. Mol. Phylogenet. Evol. 65, 339–344 (2012).
Glockner, G., Rosenthal, A. & Valentin, K. The structure and gene repertoire of an ancient red algal plastid genome. J. Mol. Evol. 51, 382–390 (2000).
Kong, F., Sun, P., Cao, M., Wang, L. & Mao, Y. Complete mitochondrial genome of Pyropia yezoensis: reasserting the revision of genus Porphyra. Mitochondrial DNA 10.3109/19401736.2013.803538.
Burger, G., Saint-Louis, D., Gray, M. W. & Lang, B. F. Complete sequence of the mitochondrial DNA of the red alga Porphyra purpurea. Cyanobacterial introns and shared ancestry of red and green algae. Plant Cell 11, 1675–1694 (1999).
Mao, Y., Zhang, B., Kong, F. & Wang, L. The complete mitochondrial genome of Pyropia haitanensis Chang et Zheng. Mitochondrial DNA 23, 344–346 (2012).
Hwang, M. S., Kim, S.–O., Ha, D.–S., Lee, J. E. & Sang-Rae, L. Complete sequence and genetic features of the mitochondrial genome of Pyropia tenera (Rhodophyta). Plant Biotechnol. Rep. 10.1007/s11816-013-0281-4 (2013).
Kucera, H. & Saunders, G. W. A survey of Bangiales (Rhodophyta) based on multiple molecular markers reveals cryptic diversity. J. Phycol. 48, 869–882 (2012).
Hus, H. T. A. Preliminary notes on west-coast Porphyras. Zoe 5, 61–70 (1900).
Conway, E. An examination of the original specimens of Porphyra perforata J. Ag. (Rhodophyceae, Bangiales). Phycologia 13, 173–177 (1974).
Conway, E., Mumford, T. F. & Scagel, R. F. The genus Porphyra in British Columbia and Washington. Syesis 8, 185–244 (1975).
Mumford, T. F. & Cole, K. M. Chromosome numbers for fifteen species in the genus Porphyra (Bangiales, Rhodophyta) from the west coast of North America. Phycologia 16, 373–377 (1977).
Cole, K. M. in Biology of the Red Algae. (eds Cole K. M., Sheath R. G.) 73–101 (Cambridge University Press, 1990).
Bebber, D. P. et al. Herbaria are a major frontier for species discovery. Proc. Natl. Acad. Sci. USA. 107, 22169–22171 (2010).
Field, D. et al. Open software for biologists: from famine to feast. Nat. Biotechnol. 24, 801–803 (2006).
Zerbino, D. R. & Birney, E. Velvet: algorithms for de novo short read assembly using de brujin graphs. Genome Res. 18, 821–829 (2008).
Namiki, T., Tanaka, H. & Sakakibara, Y. Metavelvet: an extension of velvet assembler to de novo metagenome assembly from short sequence reads. Nucleic Acids Res. 40, 1–12 (2012).
Schattner, P., Brooks, A. N. & Lowe, T. M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, 686–689 (2005).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108 (2007).
Darling, A. E., Mau, B. & Perna, N. T. ProgressiveMauve: multiple genomic alignment with gene gain, loss and rearrangement. PLoS One 5, e11147 (2010).
Katoh, K. & Standley, D. M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 30, 772–780 (2013).
Waterhouse, A. M., Procter, J. B., Martin, D. M. A. & Barton, G. J. Jalview version 2 - a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 (2009).
Stamatakis, A. RAxML Version 8: A tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 30, 1312–1313 (2014).
Goecks, J., Nekrutenko, A., Taylor, J. & The Galaxy Team. Galaxy: a comprehensive approach for supporting accessible, reproducible and transparent computational research in the life sciences. Genome Biol. 11, R86 (2010).
Blankenberg, D. et al. Galaxy: a web-based genome analysis tool for experimentalists. Current Protocols in Molecular Biology 89, 19.10.1–19.10.21 (2010).
Giardine, B. et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 15, 1451–1455 (2005).
Dereeper, A. et al. Phylogeny. fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36, W465–469 (2008).
Deng, W. et al. DIVEIN: a web server to analyze phylogenies, sequence divergence, diversity and informative sites. Biotechniques 48, 405–408 (2010).
Acknowledgements
We wish to especially thank Dr. Per Lassen, Dr. Patrik Frödén and Dr. David Giblin for sending fragments and photos of type materials, as well as Dr. Bob Waaland for his assistance in securing material. Drs. Megan Dethier and Charles O'Kelly kindly supplied fresh Pyropia from San Juan Island. Dr. Rajinder Kaul provided valuable genome sequencing advice and Joni Black provided laboratory support for this project. This work was possible as a result of the generous support from a private family trust from P.W.G. and Dr. Kelly Locke, Title V grant director at Hartnell College (Grant number- PO31C110168).
Author information
Authors and Affiliations
Contributions
J.R.H. designed, executed, coordinated and wrote the study. P.W.G. annotated, assembled and contributed to the writing of the paper. L.R. performed genomic assemblies. J.T. and M.S. annotated the data. E.R. performed the contamination analyses. C.M. and J.D.Y. executed assemblies and provided technical expertise. K.A.M. supplied specimens and contributed to the text of the paper. All authors discussed the results and commented on the manuscript. The views and conclusions contained herein are those of the authors and should not be interpreted as necessarily representing the official policies or endorsements, either expressed or implied of the US government.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Table 1
Supplementary Information
Table 2
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images in this article are included in the article's Creative Commons license, unless indicated otherwise in the image credit; if the image is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the image. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Hughey, J., Gabrielson, P., Rohmer, L. et al. Minimally destructive sampling of type specimens of Pyropia (Bangiales, Rhodophyta) recovers complete plastid and mitochondrial genomes. Sci Rep 4, 5113 (2014). https://doi.org/10.1038/srep05113
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05113
This article is cited by
-
Utility of taxon-specific molecular markers for the species identification of herbarium specimens: an example from Desmarestia japonica (Phaeophyceae, Desmarestiales) in Korea
Fisheries and Aquatic Sciences (2018)
-
The first plastid genome of a filamentous taxon ‘Bangia’ sp. OUCPT-01 in the Bangiales
Scientific Reports (2018)
-
Complete chloroplast genome of Gracilaria firma (Gracilariaceae, Rhodophyta), with discussion on the use of chloroplast phylogenomics in the subclass Rhodymeniophycidae
BMC Genomics (2017)
-
Parallel evolution of highly conserved plastid genome architecture in red seaweeds and seed plants
BMC Biology (2016)
-
Mitogenomes from type specimens, a genotyping tool for morphologically simple species: ten genomes of agar-producing red algae
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
